1. Heatmap이란?
Heatmap(히트맵)은 데이터 행렬을 색상으로 표현하여 값의 패턴을 시각적으로 쉽게 파악할 수 있는 방법입니다. 유전자 발현 데이터 분석에서 흔히 사용되며, 특정 조건에서 유전자들의 발현 변화 패턴을 확인할 수 있습니다.
Heatmap 생성 코드 분석
library('pheatmap')
df_Fc2 <- df_Fn
rownames(df_Fc2) <- df_Fc2$name
head(df_Fc2)
df_Fc2 <- df_Fc2[c("Control1","Control2","Control3","Sub1","Sub2","Sub3")]
df_Fc2 <- t(apply(df_Fc2, 1, function(x)(x-mean(x))/(max(x)-min(x))))
head(df_Fc2)
p <- pheatmap(df_Fc2, angle_col = 0)코드 설명
- df_Fn 데이터를 df_Fc2로 복사
- 행 이름을 유전자 이름(name)으로 설정
- 특정 샘플(Control1, Control2, ...)만 선택하여 데이터프레임 갱신
- 각 유전자의 발현 값을 min-max 정규화하여 -1~1 범위로 조정
- pheatmap 패키지를 사용해 히트맵 생성
아래와 같이 유전자 발현 양상을 한 눈에 볼 수 있습니다.

2. GO(Gene Ontology) 분석
GO 분석은 유전자들이 생물학적 과정(Biological Process), 세포 구성(Cellular Component), 분자 기능(Molecular Function)에 어떻게 관련되는지를 조사하는 방법입니다.
- BP (Biological Process): 생명 활동과 관련된 과정 (예: 세포 분열, 면역 반응)
- CC (Cellular Component): 유전자가 기능하는 세포 구조 (예: 미토콘드리아, 핵)
- MF (Molecular Function): 유전자가 수행하는 분자적 기능 (예: 단백질 결합, 효소 활성)
GO Plot 생성 코드 분석
### BP
dt_GO1 <- read.table("GO_BP.txt", sep="\t")
names(dt_GO1) <- as.matrix(dt_GO1[1,])
dt_GO1 <- dt_GO1[-1,]
dt_GO1['Count'] <- as.numeric(as.character(unlist(dt_GO1['Count'])))
dt_GO1['PValue'] <- as.numeric(as.character(unlist(dt_GO1['PValue'])))
df_split <- data.frame(do.call('rbind',
strsplit(as.character(dt_GO1$Term),
split = '~', fixed = TRUE)))
dt_GO1$BP <- df_split$X2
dt_GO1_BP <- dt_GO1[c("Count", "BP", "PValue")]코드 설명
- GO_BP.txt 파일을 읽어 데이터프레임으로 변환
- 열 이름을 설정하고 첫 행 제거
- Count와 PValue 값을 숫자로 변환
- Term 열을 ~ 기준으로 분리하여 BP 컬럼 생성
- 최종적으로 Count, BP, PValue만 포함된 데이터프레임 생성
데이터는 아래에 나오는 방식으로 받아 활용할 수 있습니다.
2024.07.08 - [생물정보학] - DAVID - gene 관계 분석 수행
DAVID - gene 관계 분석 수행
[DAVID: Functional Annotation Bioinformatics Microarray Analysis] DAVID (Database for Annotation, Visualization and Integrated Discovery)는 연구자들이 대규모 유전자 목록의 생물학적 의미를 이해할 수 있도록 돕는 포괄적인
s-story-world.tistory.com
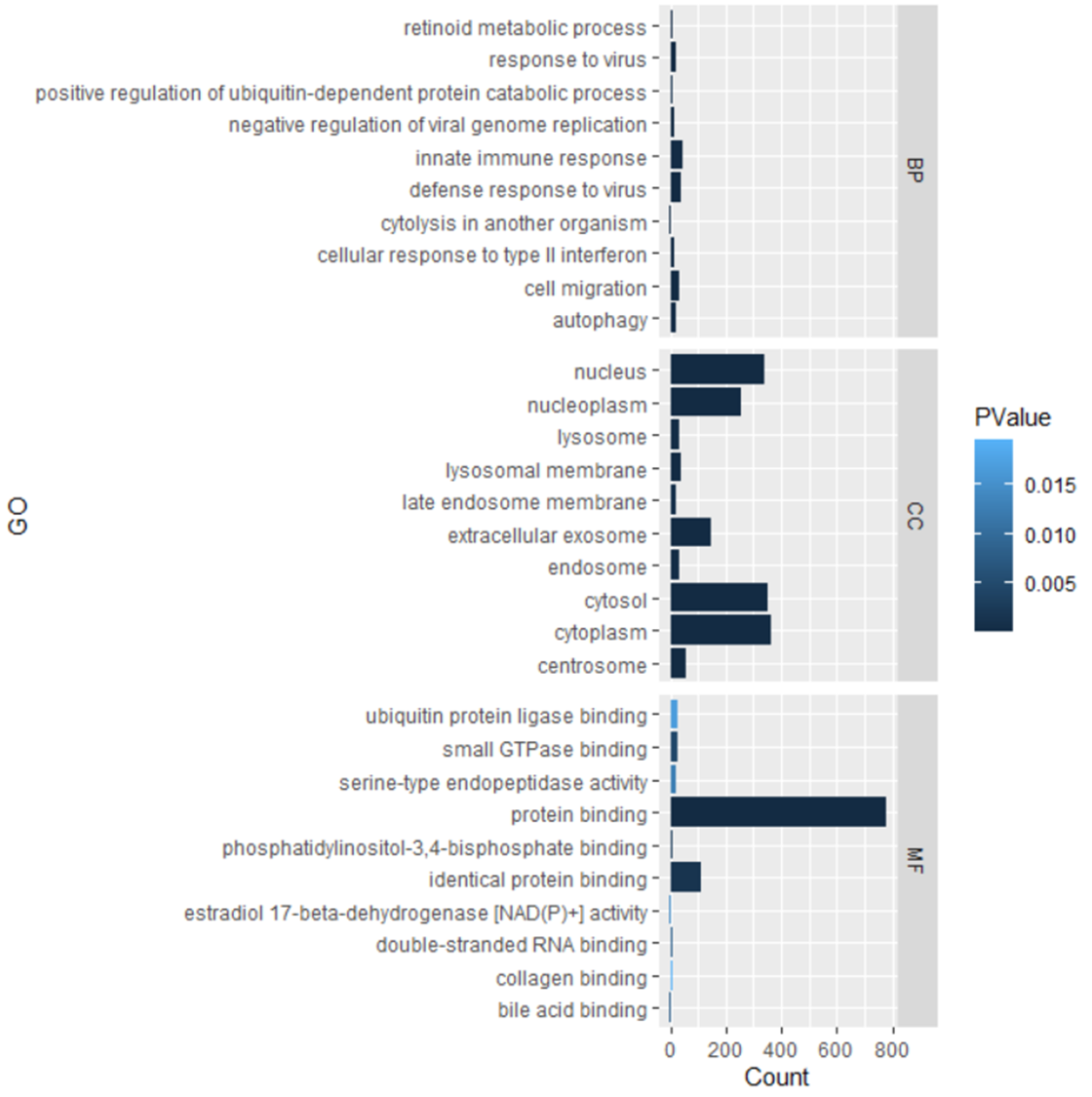
GO Plot 시각화
dt_GO1_BP$grid <- "BP"
colnames(dt_GO1_BP) <- c("Count", "GO", "PValue", "gird")
dt_GO1_CC$grid <- "CC"
colnames(dt_GO1_CC) <- c("Count", "GO", "PValue", "gird")
dt_GO1_MF$grid <- "MF"
colnames(dt_GO1_MF) <- c("Count", "GO", "PValue", "gird")
dt_GO1 <- rbind(rbind(dt_GO1_BP[1:10,], dt_GO1_CC[1:10,]), dt_GO1_MF[1:10,])
ggplot(data = dt_GO1) + geom_bar(aes(x=Count, y=GO, fill=PValue), stat="identity") +
facet_grid(vars(gird), scales="free_y")코드 설명
- BP 데이터를 ggplot2 패키지를 사용해 시각화
- facet_grid()를 사용해 BP, CC, MF를 구분하여 시각화
- fill=PValue를 설정하여 색상으로 p-value 표현
3. KEGG Pathway 분석
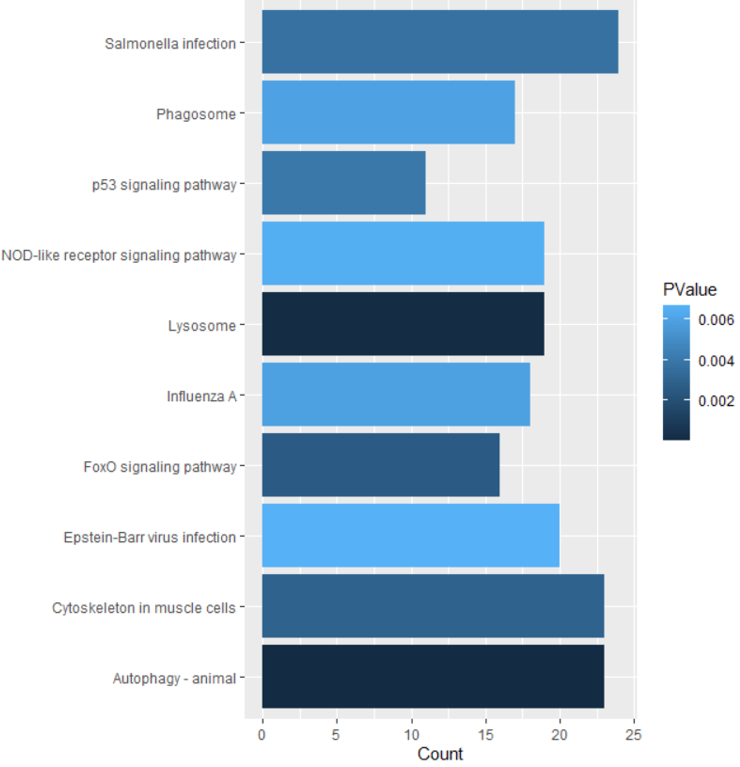
KEGG(Kyoto Encyclopedia of Genes and Genomes) 분석은 유전자들이 어떠한 생물학적 경로(Pathway)에 관여하는지를 분석하는 방법입니다. 특정 기능을 가진 유전자들이 네트워크를 형성하며, 신호 전달 경로나 대사 과정 등을 연구할 때 사용됩니다.
KEGG Plot 생성 코드 분석
dt_GO1 <- read.table("KEGG.txt", sep="\t")
names(dt_GO1) <- as.matrix(dt_GO1[1,])
dt_GO1 <- dt_GO1[-1,]
dt_GO1['Count'] <- as.numeric(as.character(unlist(dt_GO1['Count'])))
dt_GO1['PValue'] <- as.numeric(as.character(unlist(dt_GO1['PValue'])))
df_split <- data.frame(do.call('rbind',
strsplit(as.character(dt_GO1$Term),
split = ':', fixed = TRUE)))
dt_GO1$Term2 <- df_split$X2
library(ggplot2)
ggplot(data=dt_GO1[1:10,], aes(x=Count, y=Term2, fill=PValue)) +
geom_bar(stat = "identity")코드 설명
- KEGG.txt 파일을 불러와 데이터프레임으로 변환
- Count와 PValue를 숫자로 변환
- Term을 : 기준으로 분리하여 Term2 생성
- ggplot2 패키지를 활용해 상위 10개의 KEGG pathway를 막대 그래프로 시각화
데이터는 아래에 나오는 방식으로 받아 활용할 수 있습니다.
2024.07.08 - [생물정보학] - DAVID - gene 관계 분석 수행
DAVID - gene 관계 분석 수행
[DAVID: Functional Annotation Bioinformatics Microarray Analysis] DAVID (Database for Annotation, Visualization and Integrated Discovery)는 연구자들이 대규모 유전자 목록의 생물학적 의미를 이해할 수 있도록 돕는 포괄적인
s-story-world.tistory.com
'R note' 카테고리의 다른 글
| R 데이터 프레임에서 특정 열 값 변환하기 (0) | 2025.03.13 |
|---|---|
| R에서 t.test() 오류 해결: "데이터는 본질적으로 상수입니다" (0) | 2025.03.10 |
| miRNA 데이터 분석 및 시각화 (0) | 2025.02.01 |
